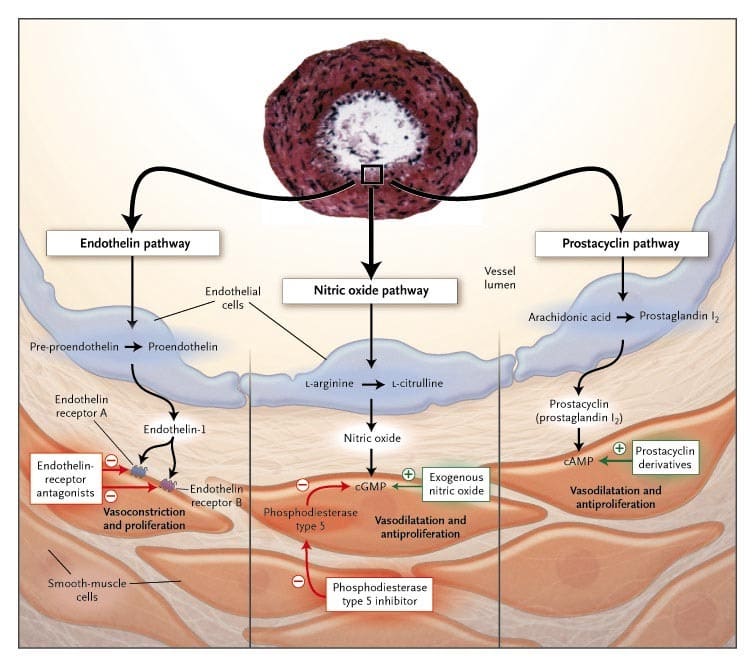
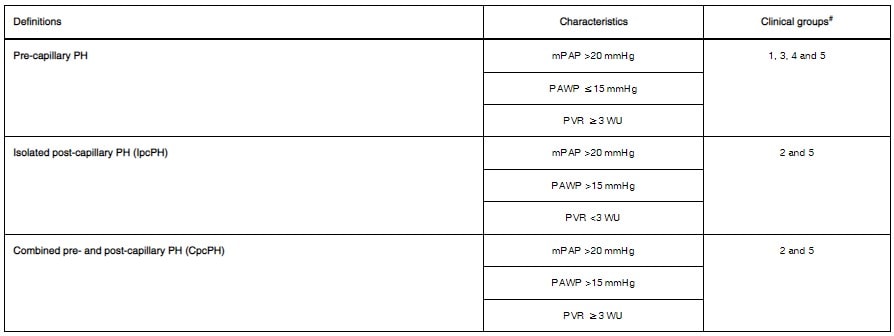
- Pre-capillary vs Post-capillary PH determined based on right heart catheterization
{kind=link}
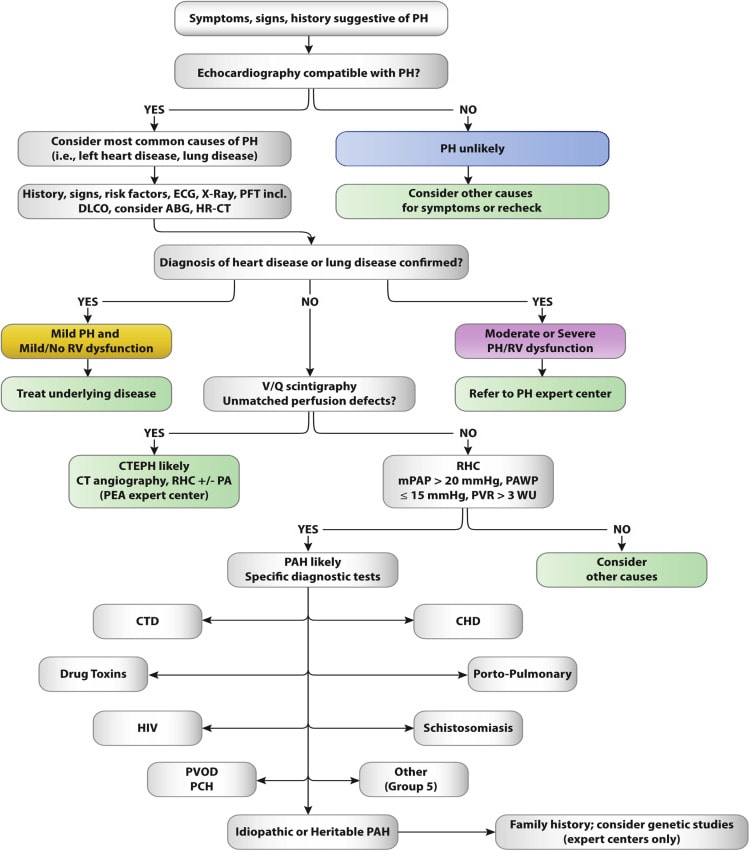
- PAH must be diagnosed by excluding other more common etiologies (left heart disease, lung disease, and CTEPH)
- Vasoreactivity testing in select PAH patients as mentioned above
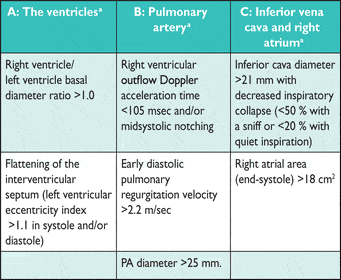
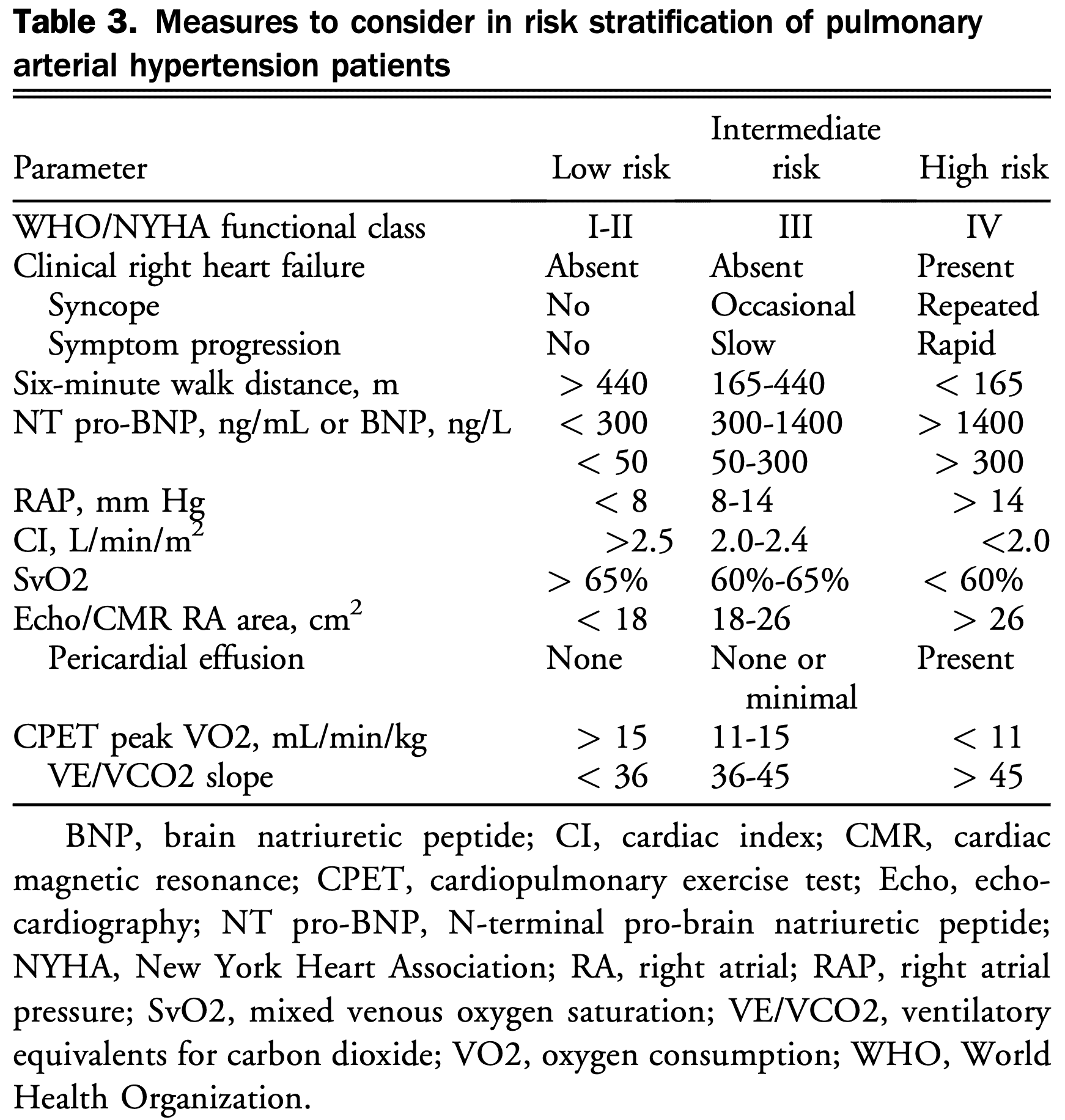
- Risk stratification using a combination of clinical, hemodynamic, and imaging features
{kind=link}